
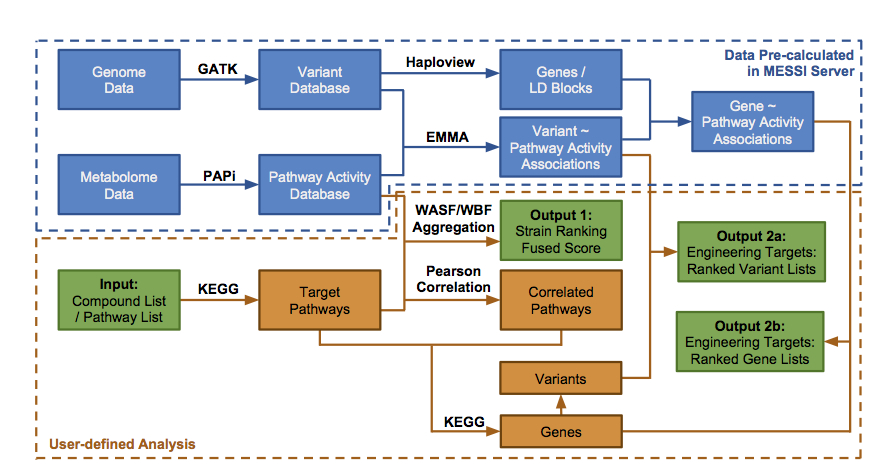
The workflow of MESSI is illustrated below:
Job ID is generated automatically by the access time, but the user can choose any unique ID easier to memorize. Since users are not requested to log in, completed jobs could be reviewed only by job IDs.
Datasets compatible with MESSI are expected to encompass metabolomic data from large-scale genetic studies. The first available database was named DB01_SC_21, including 21 S. cerevisiae strains. In the databases' detail page, strains and their sources (fermentation, clinical, baking or lab strain) are listed (example below).

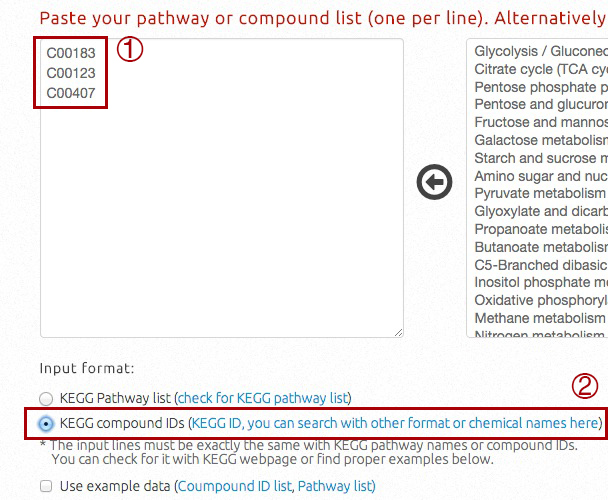
Two kinds of input formats are acceptable in MESSI and these are KEGG pathway names and KEGG compound IDs. Only one format would be processed in each job. Examples for both types of input data are available for testing usage. When "Use example data" is selected, user-defined input will be ignored.
When input is a pathway list, users should:
i) select the input pathways from a candidate list on the web server (use Ctrl, Shift or Cmd to select multiple pathways);
ii) press the left arrow to add selected pathways into the input box;
iii) directly type pathway names in the input box, or make mondification on existing ones;
iv) select "KEGG Pathway list" (default option) as input format.

When input is compound ID list, users should:
i) directly type compound IDs in the input box;
ii) select "KEGG Compound IDs" as input format.
Variants and genes associated with input pathways or compounds could be summarized and demonstrated in different biological levels, serving as potential metabolic engineering targets. When a higher level is chosen, data from lower levels are automatically processed. Different levels are optional and include:
i) level 0, no variant and gene information will be processed (defult option);
ii) level 1, variants and genes in metabolic reactions of target compounds (only when input is a compound list);
iii) level 2, variants and genes in target pathways associated with their pathway activity scores;
iv) level 3, variants and genes in correlated pathways (click for more details) associated with their activity scores;
v) level 4, variants and genes in whole genome level associated with activity in target pathways.

Variants (SNPs and InDels) could be filtered optionally according to their mutation effects/positions, including upstream (800bp region) non-coding variants, synonymous coding (including synonymous amino acid coding codon or synonymous stop codon), missense coding, nonsense variants (stop-codon gained, start- or stop-codon lost situations), frameshifts, codon insertions or deletions, intron, and variants in splice sites.

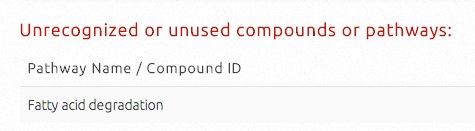
Pathways or compounds may be markeed "Unrecognized or Unused" and will not be processed in pathway activity score calculation, strain ranking and metabolic engineering targets prediction. There are four possibilities that pathways or compounds may be marked as “unrecognized or unused compounds or pathways”:
i) their input IDs or names could not be recognized by KEGG;
ii) the input IDs or names are recognized but are not part of the S. cerevisiae KEGG database;
iii) no pathway activity scores could be calculated due to data incompleteness in the metabolomic profiling of these pathways;
iv) for recognized compounds, pathway activity scores are not available for all associated pathways (similar to (iii)).
The example shown below is due to (iii).
In this part, users can set "Expectation", "Weight", "Call Variants" for each recogonized pathway, and select the pathway activity aggregation algoritm.
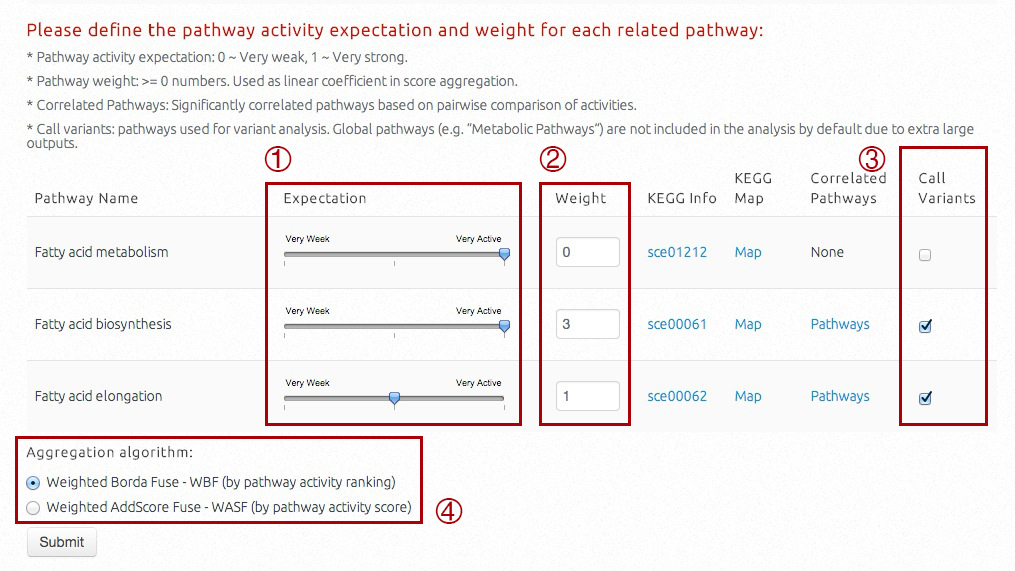
Expectation is to characterize how active the target pathway should be. Three options are available including Very Active, Medium and Very Weak by setting the slider.
Weight is to characterize how important is a pathway compared to others in the final ranking of strains. It can be any non-negative value used as folds during aggregation. Weight for universal pathways is set to 0 by default.
Hyperlinks to KEGG information and KEGG maps are provided.
If users would like to process metabolic engineering targets in correlated pathways or whole-genome level, pathways correlated with specific target pathway will be shown. By clicking "Pathways", the table will include name and ID of target pathway, names and IDs of correlated pathways, Pearson correlation coefficient (-1~0, negative correlation, 0~+1, possitive correlation), and correlation P-values and FDR adjusted Q-values.
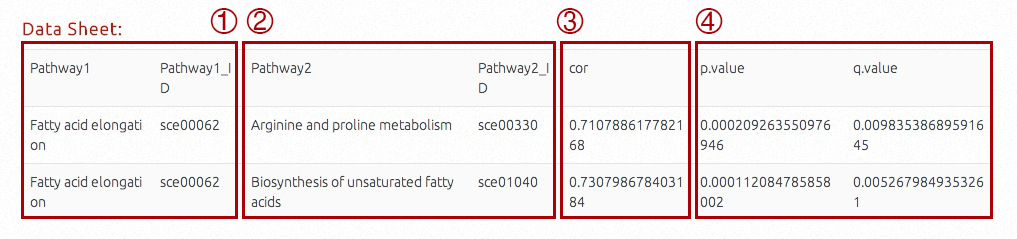
The check box should be checked if metabolic engineering targets (variants and genes) are supposed to be presented for this target pathway. Similarly, this option is not used for universal pathways by default.
User can also select two optional pathway activity aggregation algorithms, WBF and WASF. In respective primary ranking, WBF only considers activity-based rankings, while WASF uses the actual scores.
Job information including job ID, input type, database and parameters is provided.

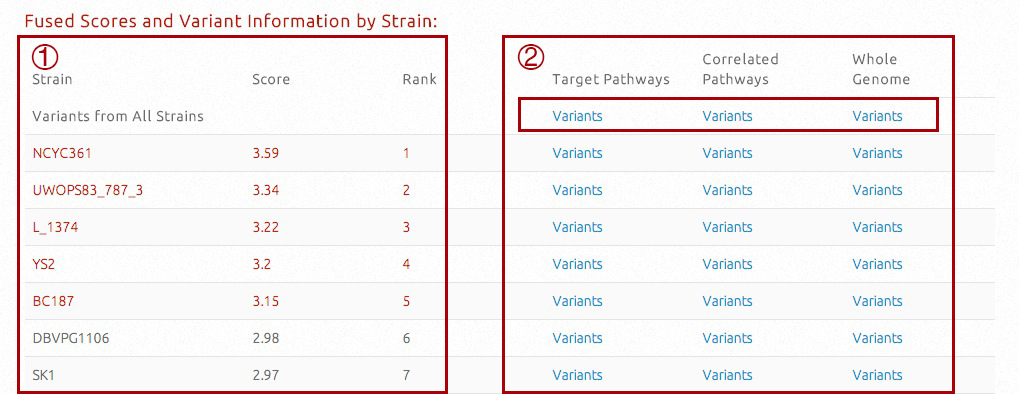
Based on the pathways' fused activity scores, the best strains (top 5) are identified.

This part includes the resluts for metabolic engineering target prediction.
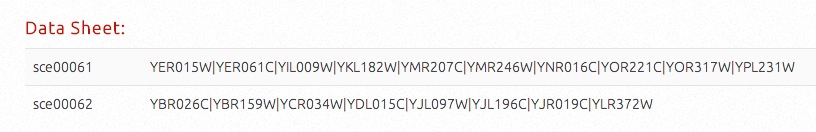
MESSI incorporates information of compounds, pathways and genes and their relationships from KEGG database using KEGG API REST. Pathway correlations are calculated by Pearson correlation of the pathway activity scores. This information will be shown to users.

By viewing the correlations, the targets (pathway IDs or compound IDs) will be listed in the first column and related items (pathway IDs or gene IDs) will be listed in the second coloumn, seperated by '|'.
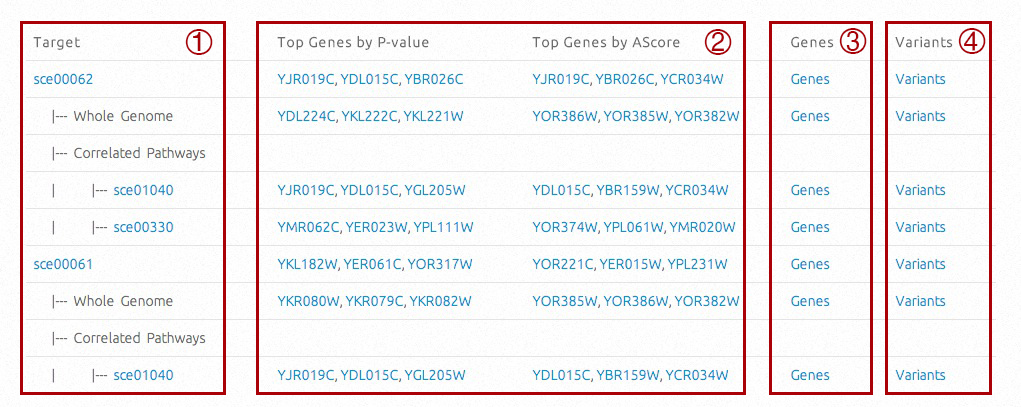
Target compounds or pathways and correlated pathways will be listed in a tree structure, with hyperlinks to relative KEGG information. Correlated pathways and whole-genome level information will be available if selected. For pathways, the top three genes associated with their pathway activities will be listed in two columns (P-values based and A-Scores based respectively, click for more details). All affecting genes and variants will be available on the right.
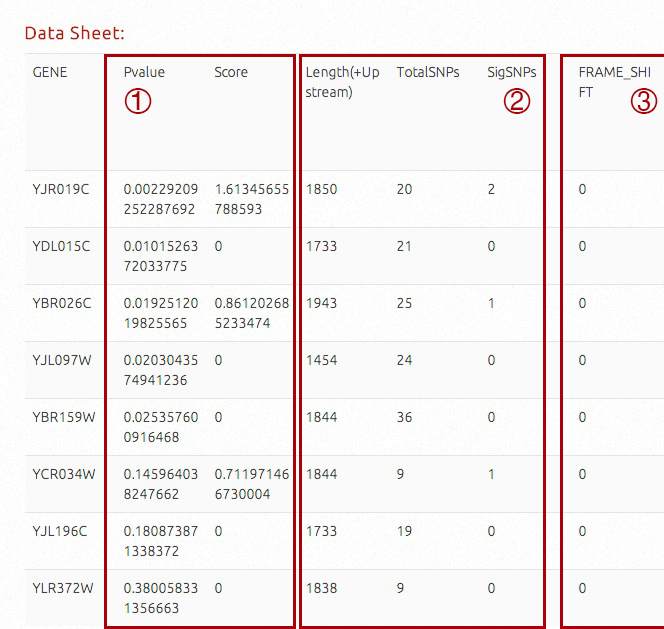
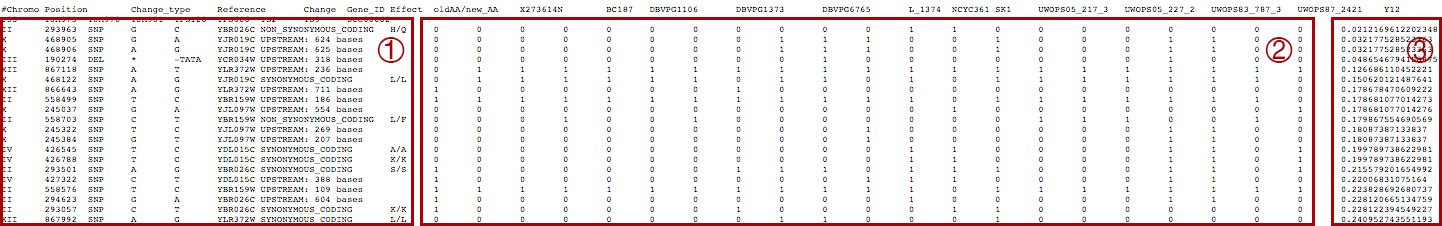
For gene-scale information, gene names, association P-values, A-Scores, gene region length (800bp upstream region included), total variant counts, significant (P < 0.05) variant counts and statistics for significant variants of different effects are available. This table is sorted by association P-values by default.
For variant-scale information, four main parts are presented including variant information (possition, allele type, effect, etc.), strain matrix (0: without this variant, 1: with this variant) and association P-values to pathway activities. When there is only one column in association P-values, this table is sorted by the P-values.
In this table, fused scores and the final ranking of all strains will be presented. Universal strain-specific variants will be presented, for different biological levels.
All results are downloadable, including job information and parameters, target pathway information with expectation and weight, strain scores and ranking, relationship among input targets, correlated pathways and genes, variant lists and gene-level statistics.

Finished jobs could be review by job IDs. After submitting, web page will be redirected to the relevant results page.
Pathway Activity Calculation: PAPi (Aggio, Ruggiero & Villas-Boas, 2010)
Pathway Activity Aggregation: WASF/WBF (Saari, 1999)
Pathway Correlation: Pearson Correlation with Bonferroni Correction
Association Analysis of Variants and Pathway Activities: EMMA (Kang et al, 2008)
Association Analysis of Genes and Pathway Activities: Haploview + A-Score
P-value: gene-level association P-values were calculated based on significant linkage disequilibrium (LD) blocks and proxy clusters (Hong et al, 2009). LD blocks were generated by the Haploview by applying the Four Gamete Rule (Barrett et al, 2005).
Variant significant level accumulated score (AS or A-Score) is defined by the following formula:
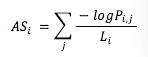
Where Li stands for the length of the gene i region (800 bp upstream sequence is included as potential regulatory region), and Pi,j stands for the P-value for significant variant j in gene i.
Kang, K., Li, J., Lim, B. L., & Panagiotou, G. (2015). MESSI: metabolic engineering target selection and best strain identification tool. Database, 2015, bav076.